
病史:女,61岁,骶髂关节病变。
大体所见:灰红带骨碎组织,体积:10.5 cm×8 cm×2cm。
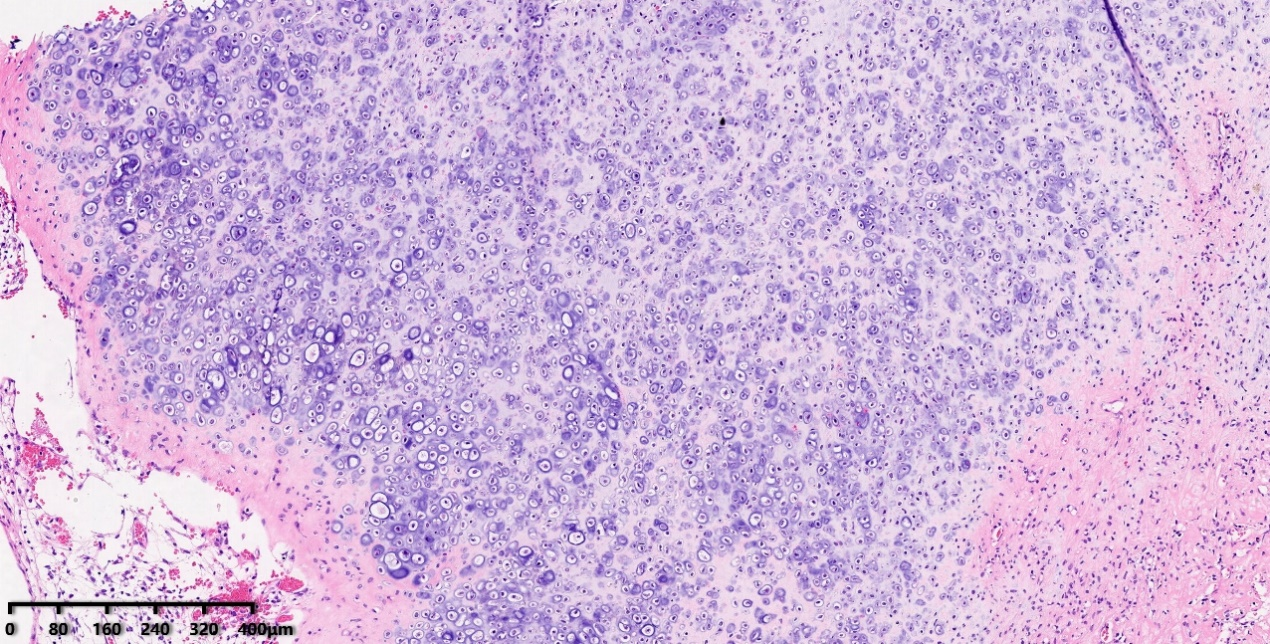
镜下特征:肿瘤细胞弥漫分布,局灶可见不同程度分化的软骨区域及鹿角样血管,肿瘤细胞胞浆稀少,胞核明显,核浆比高,染色质开放,可见一个/数个小核仁,核分裂象多见。
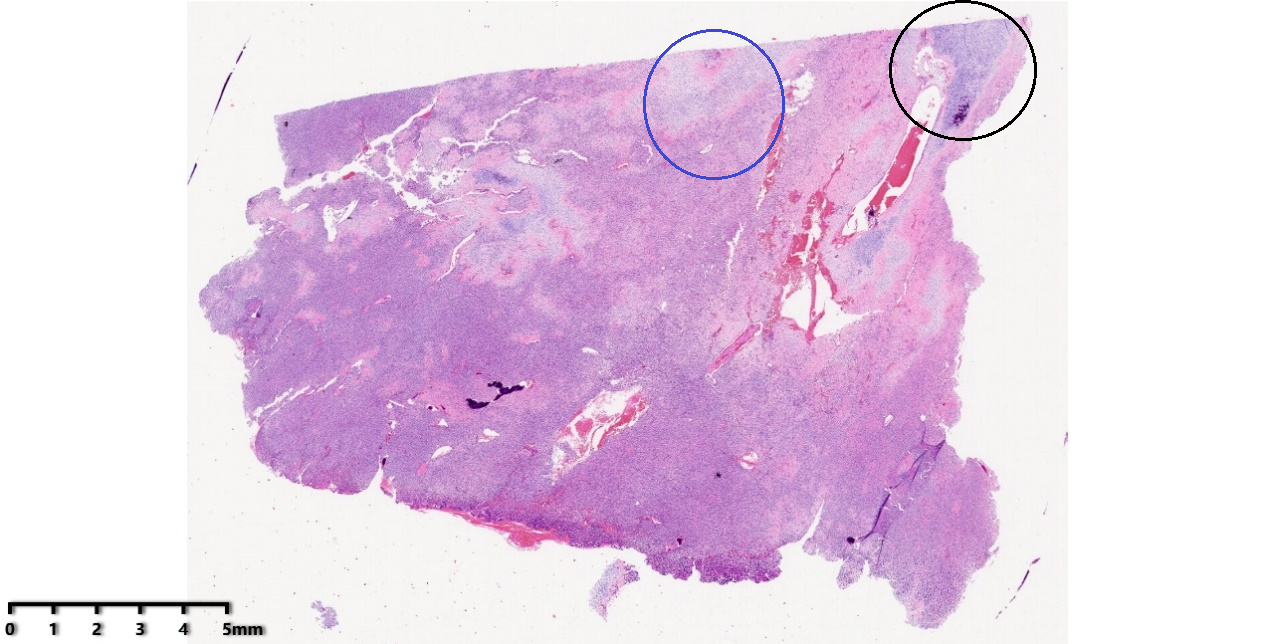
低倍镜:肿瘤细胞密度高,集中于左下方,余处可见扩张、充血的薄壁血管及散在分布的软骨岛。
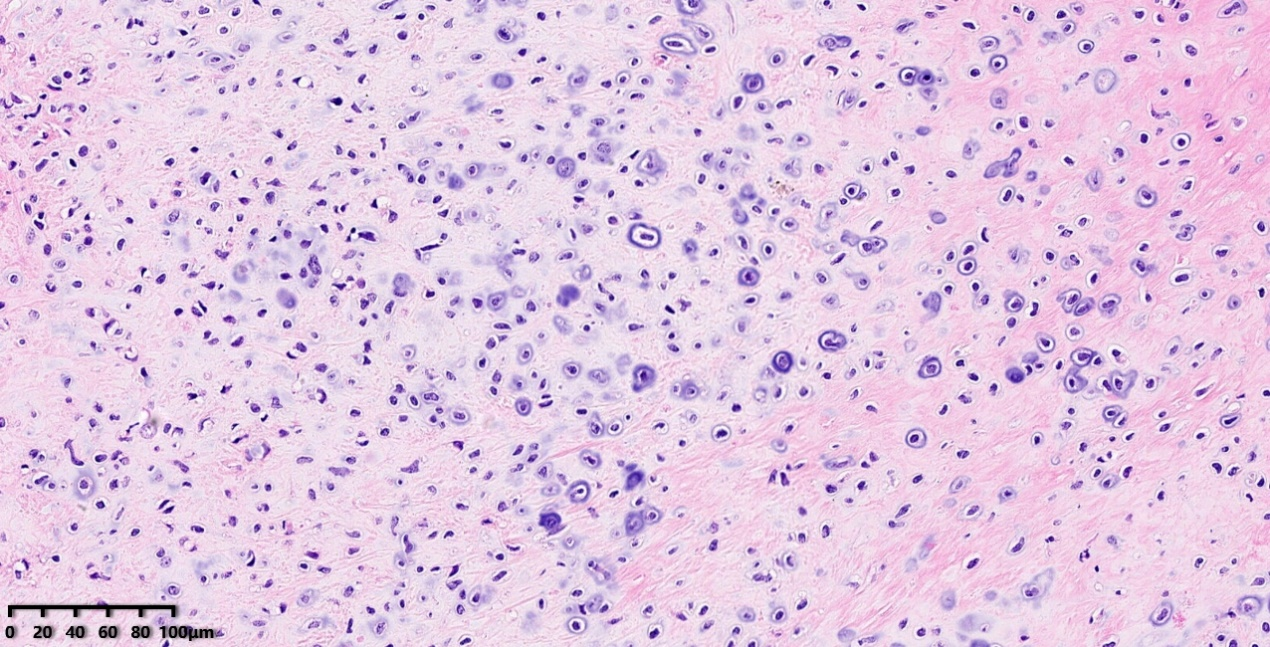
低倍镜:将上图蓝色圆圈部分放大,软骨细胞分布均匀。
高倍镜:细胞核呈圆形-椭圆形,染色质浓集,分化较成熟。
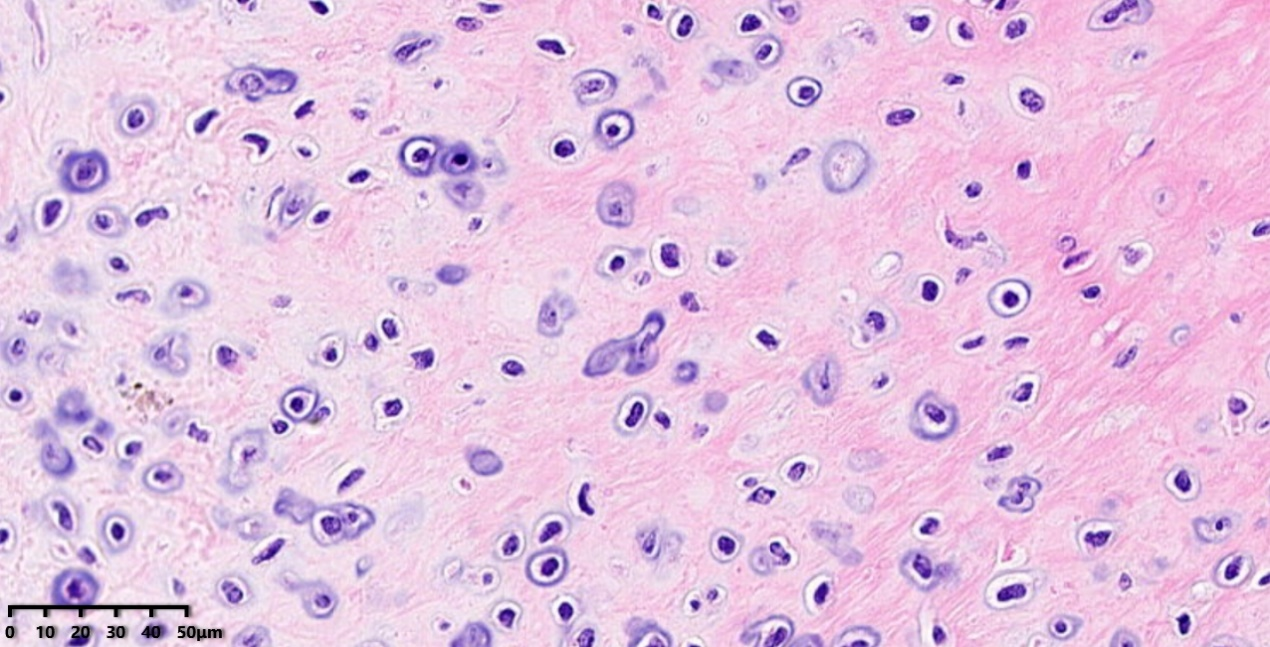
低倍镜:将上图黑色圆圈部分放大,可见软骨细胞密度明显增加。
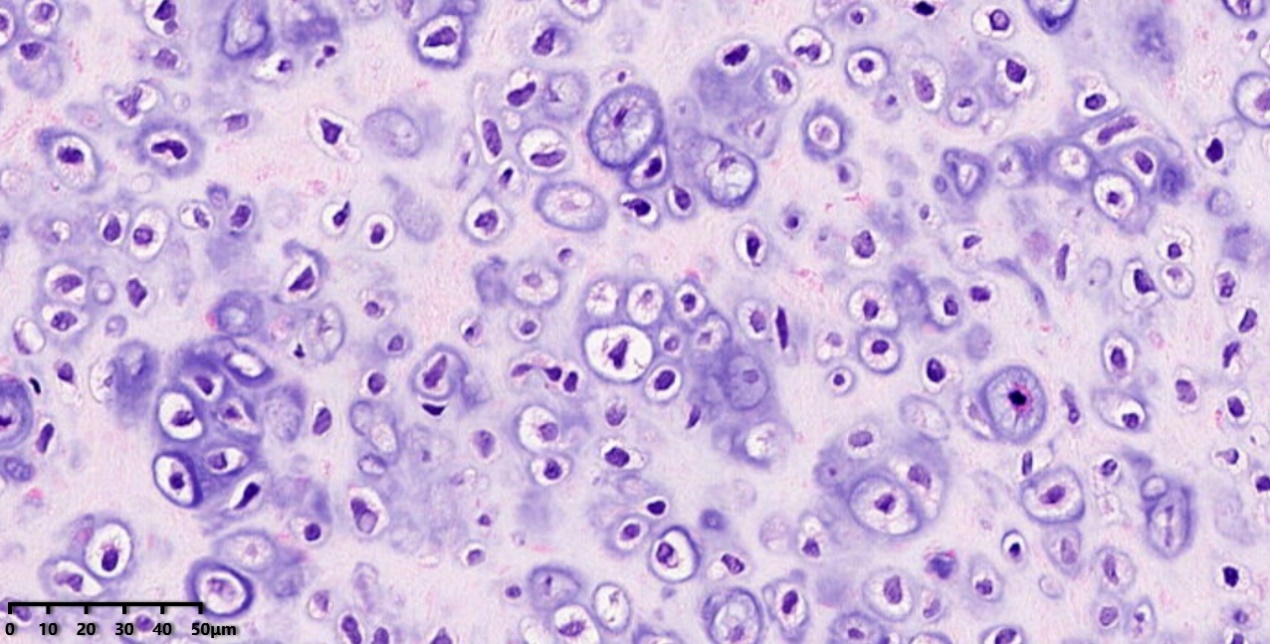
高倍镜:细胞核稍大,核型不规则,具有轻度异型。
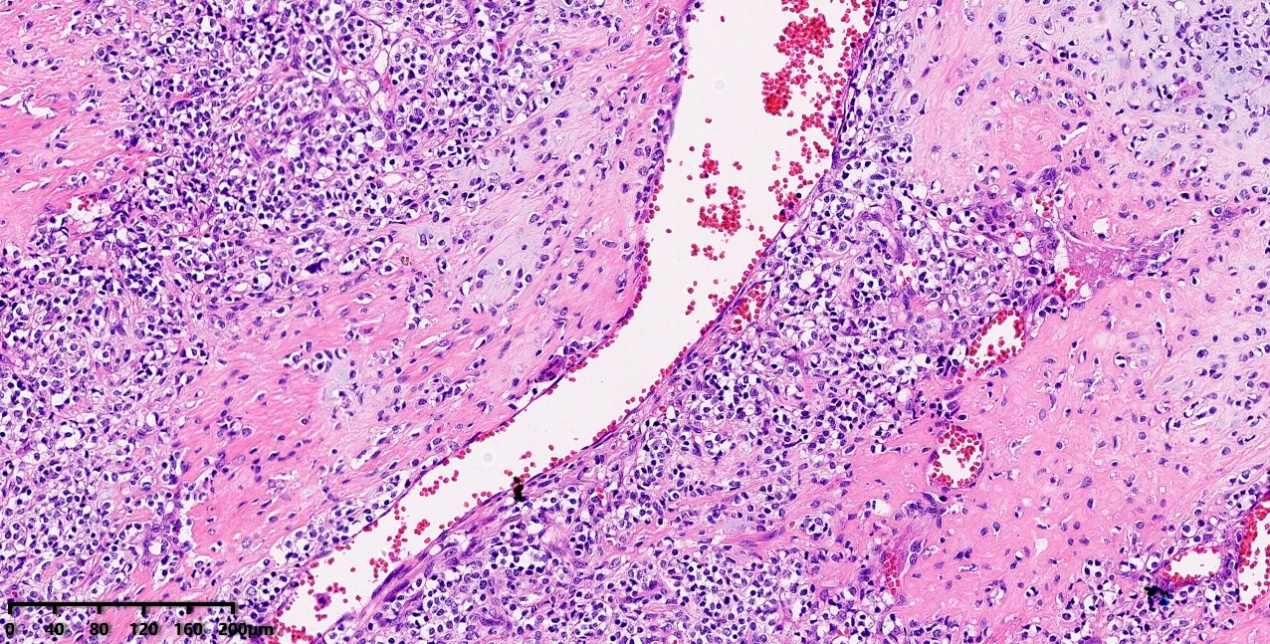
低倍镜:肿瘤细胞围绕血管呈血管外皮瘤样生长。
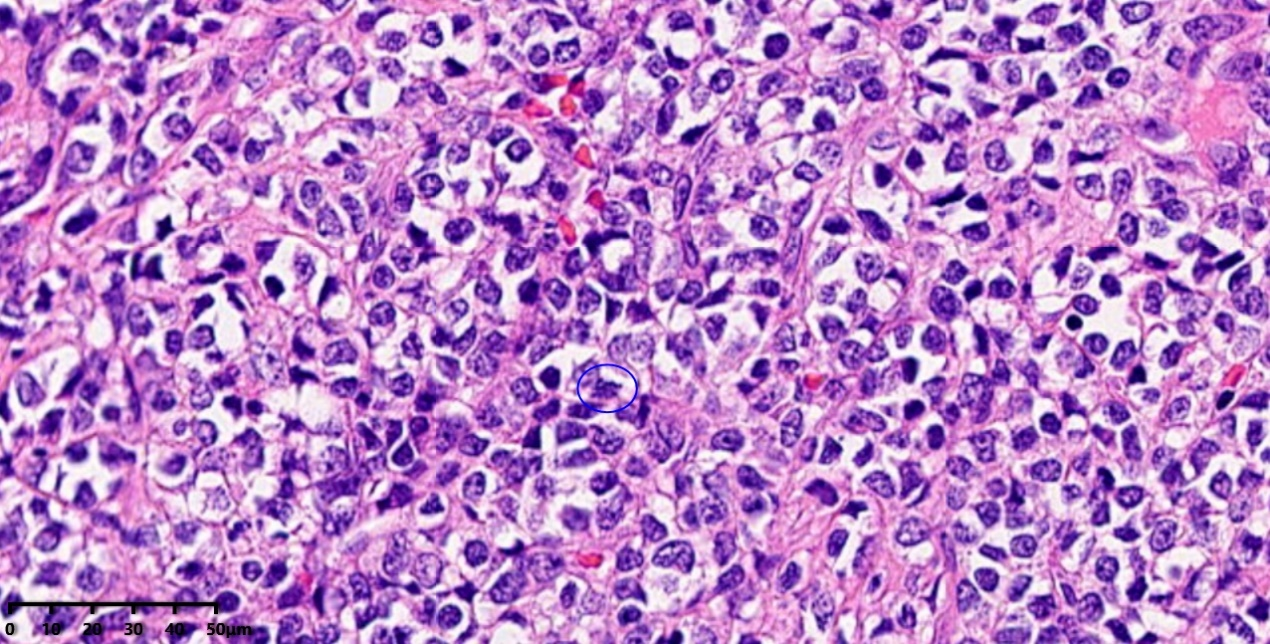
高倍镜:肿瘤细胞胞浆稀少,胞核明显,核浆比高,染色质开放,可见一个/数个小核仁,核分裂象(蓝色圆圈)可见。
免疫组化: CD99(核旁点状+)、SOX9(+)、SATB2(+)、CK(-)、LCA(-)、CD38(-)、SOX10(-)、H3 K36M(-)、H3.3 G34W(-)、SSX-CT(-)、CD38(-)、ERG(-)、CD34(-)、CgA(-)、Syn(-)、Ki-67(增殖指数约30%)。
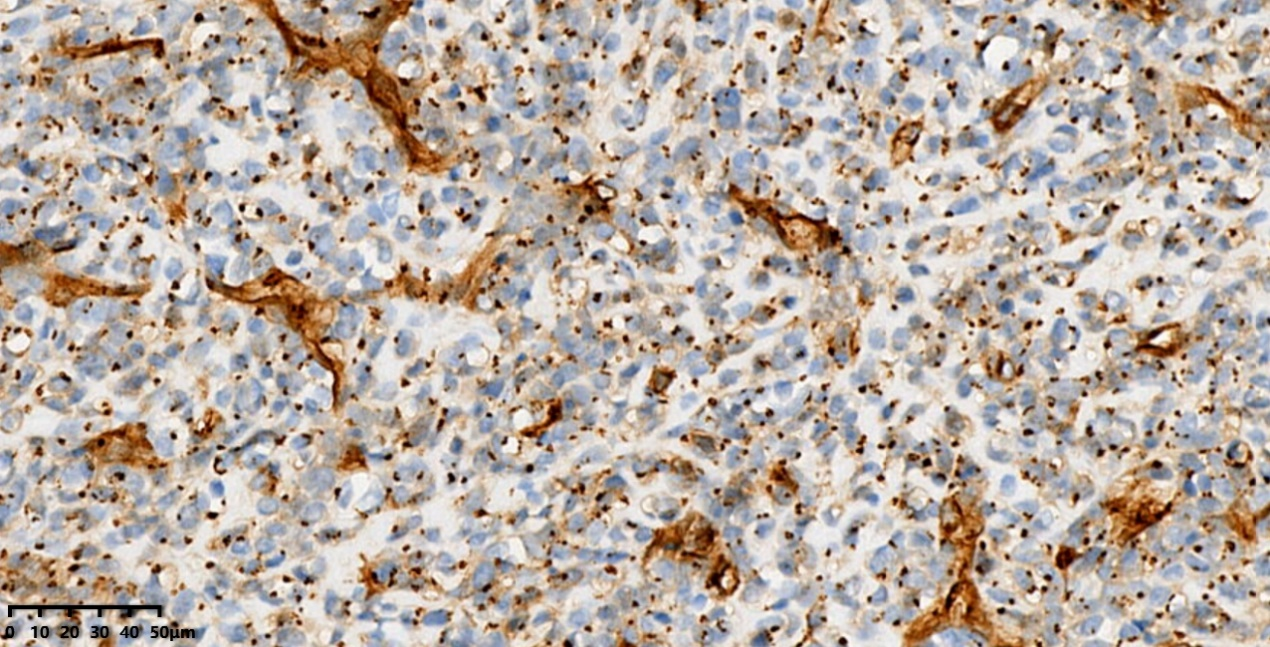
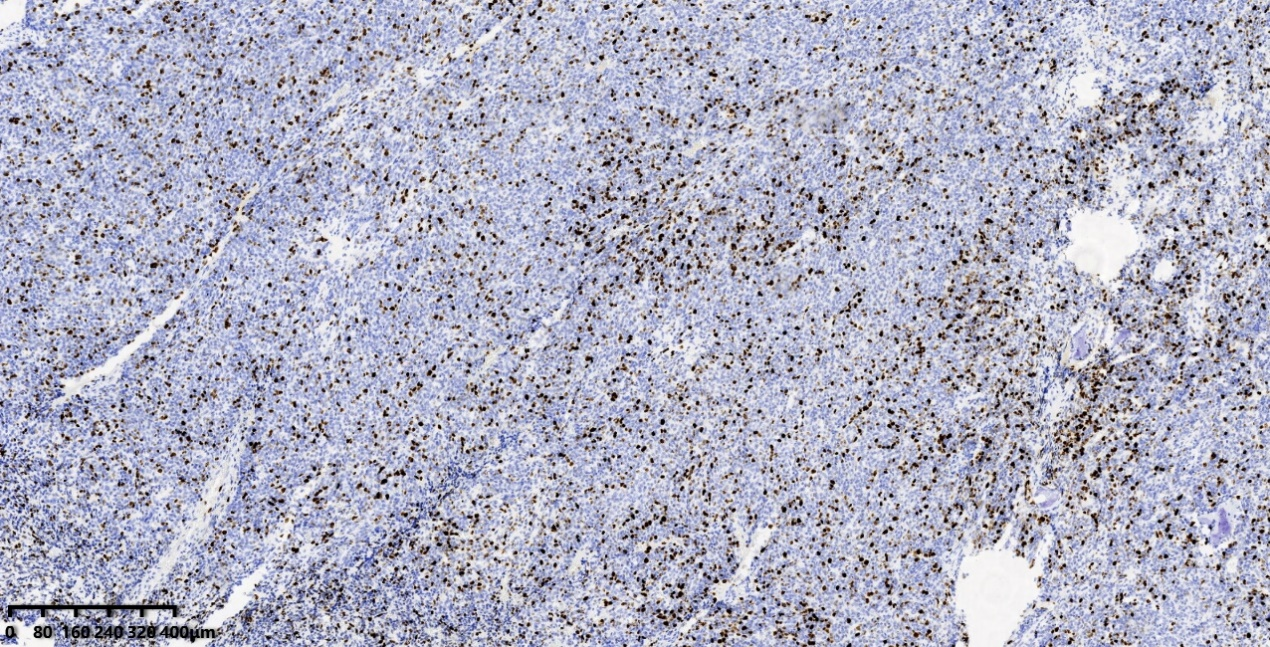
CD99
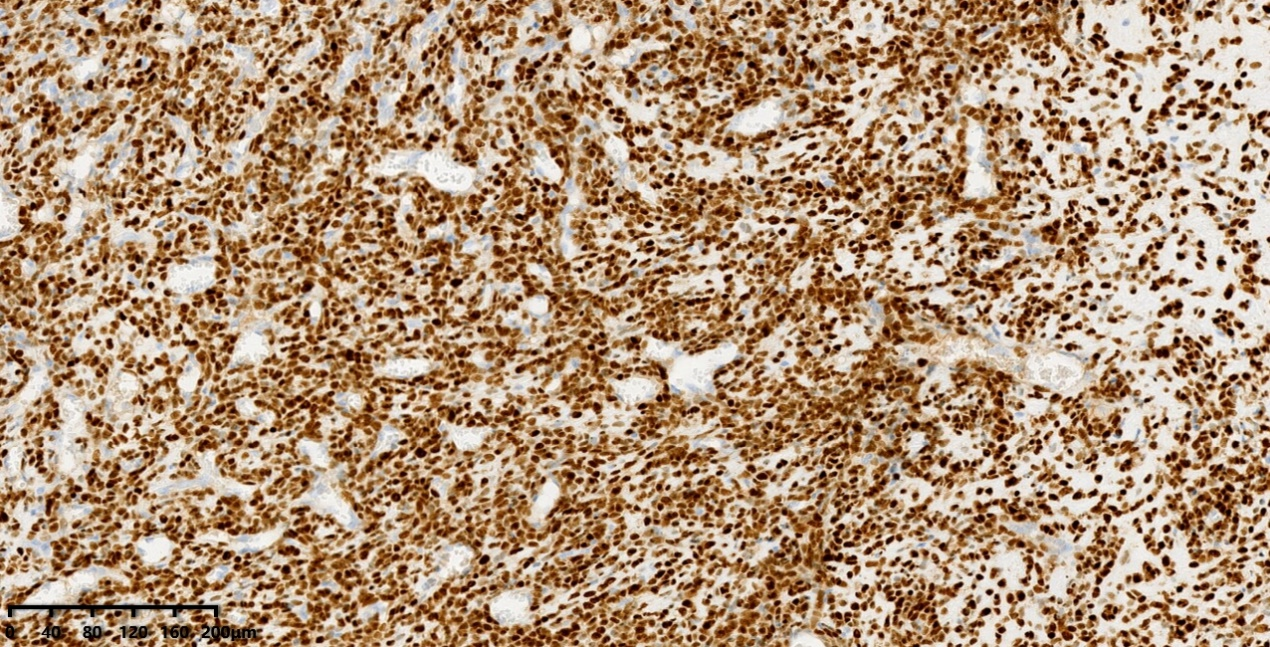
SOX9
SATB2
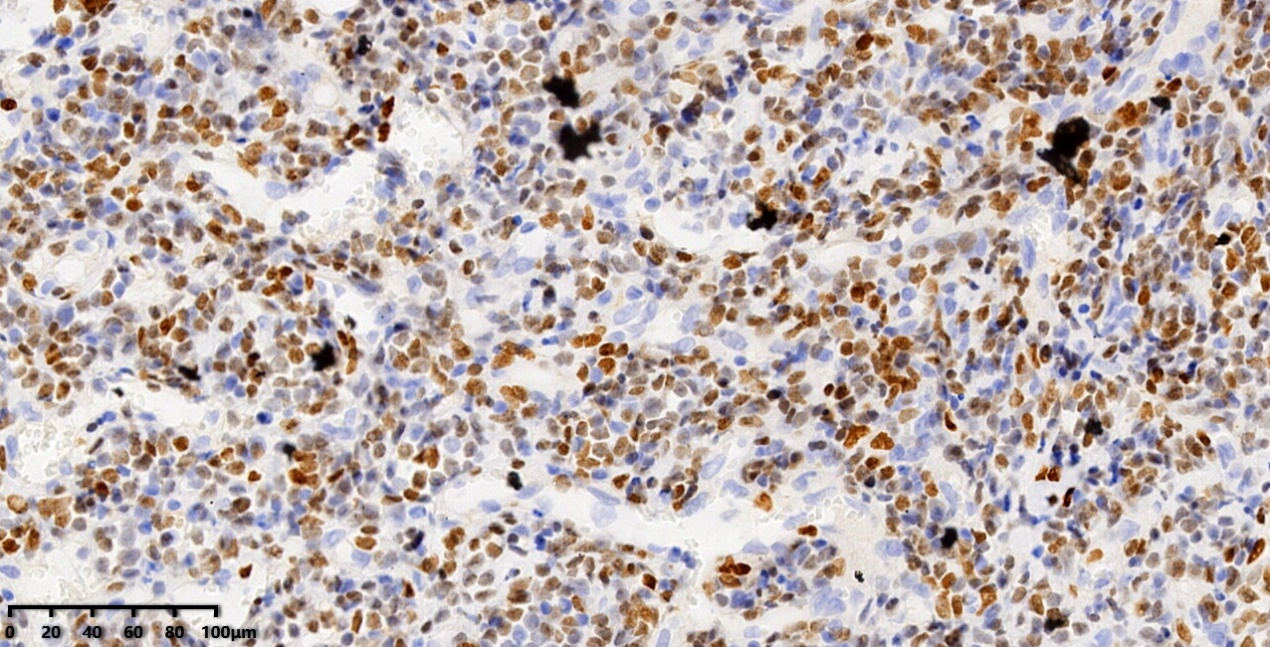
Ki-67
分子检测结果:检测到HEY1-NCOA2基因融合。
病理诊断:间叶性软骨肉瘤。
讨论
定义:一种高级别、恶性、双相性、原始间叶肿瘤,同时伴有高分化透明软骨成分。
ICD-O编码:9240/3
临床特征:主要症状为疼痛和肿胀。发病范围广,包括骨、软组织和颅内。骨内病变主要发生于颅面骨(下颌骨占 50%),约 40%发生于躯干软组织。脑膜是骨外最常见的发病部位。大约占所有软骨肉瘤的 2~4%。发病年龄范围广,高峰为 20~30 岁,平均年龄 30 岁。男性稍多(男女比 1.3:1)。
病理形态:
1、大体特征:外观多样,切面呈灰白色、白色和粉红色,质地硬或软,界限清楚。肿瘤最大径 0.9~30cm。多数病变存在钙化,范围从散在点状至片状,软骨样成分或伴有坏死和出血的片状小圆细胞都可以成为其主要成分。
2、镜下特征:肿瘤由小至中等大小、分化差的、伴有高核浆比的小圆细胞构成,存在特征性的鹿角样或血管周细胞样特征,混杂有数量不等的高分化透明软骨岛。部分区域可出现沉积的基质类似于骨样基质。。
3、免疫组化:S100、CD99、SOX9、NKX2.2、NKX3.1、IN1 阳性,FLi1 阴性。异常表达 EMA、desmin、myogenin 和 MYOD1。SMA、GFAP 和 CK 阴性。
分子遗传学特征:间叶性软骨肉瘤存在重复性的 HEY1-NCOA2 重排,其他软骨肉瘤不存在,可以作为明确的分子诊断标志物。也有报道存在 IRF2BP2-CDX1基因融合。
预后:侵袭性肿瘤,5 年和 10 年总生存率为 60%和 40%。生存率与肿瘤转移、肿瘤大小和发病部位相关,但没有与预后相关的组织学特征。儿童、青年人和年轻的成年人预后稍好。颅面部肿瘤具有惰性的临床过程,预后较好。发生于中轴骨的肿瘤预后差。化疗联合完整切除为首选治疗方法。
鉴别诊断:
1、尤文肉瘤/PNET:小圆细胞组成,呈密集/片状,小叶间有宽窄不等的纤维结缔组织间隔,可见Homer-Wright菊形团结构,无肿瘤性软骨岛形成。肿瘤细胞表达NKX2.2、CD99、Fli-1 ,90%的病例可出现EWS-FLI1基因融合。
2、去分化软骨肉瘤:无形态一致的细胞区,组织学上显示分化区与未分化区的突然转变,在低级别软骨肉瘤的基础上出现明显异型的高级别肉瘤。
3、小细胞性骨肉瘤:由圆形或短梭形细胞构成,在小细胞间有粗糙类似蕾丝样骨样组织,没有软骨成分。
4、孤立性纤维性肿瘤:以杂乱或呈席纹状的梭形细胞为主,有鹿角分支状血管,缺少软骨岛形成,STAT6、CD99、CD34在肿瘤细胞中表达,存在 NAB2-STAT6融合。
5、滑膜肉瘤:可见“血管外皮瘤”样血管结构,部分可见化生性骨化,缺乏软骨成分,此外存在SYT-SSX融合。
参考文献:
1、第5版骨与软组织WHO
2、潘梓欣,肖钦沛,孙凯璇,等.NKX3.1阳性的间叶性软骨肉瘤2例并文献复习[J].诊断病理学杂志,2023,30(12):1111-1113+1116.


 苏公网安备 32011402011742
苏公网安备 32011402011742